




SynTeam
LAFERRIERE Florent (Chercheur) DE LA SEIGLIERE Hortense (Ingénieure Technicienne ) LETOURNER Aenora (Doctorante) SABATIER Ludivine (Doctorante)
The pathological aggregation of neuronal proteins is a key and often causal event in many neurodegenerative diseases. Synucleinopathy, characterized by the accumulation and spread of aggregated amyloid forms of alpha-synuclein in the brain, is observed in Parkinson’s disease (PD), multiple system atrophy (MSA), and dementia with Lewy bodies.
Understanding the molecular mechanisms involved in the amyloid self-assembly of this small synaptic protein, tracking the cellular processes by which neurons handle its pathogenic conformational changes and deciphering the inter-neuronal propagation pathways of these aggregated forms are crucial aspects in neurodegenerative research and hold significant interest for therapeutic development.
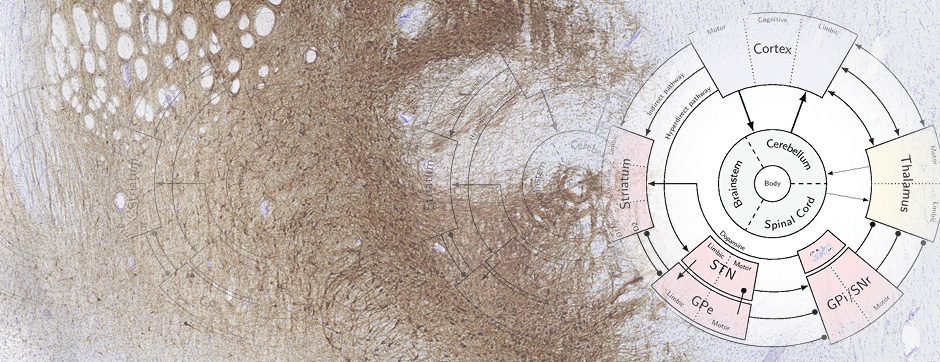
It is now well-established that aggregated alpha-synuclein can perpetuate the pathological process by amplifying its own aggregation through a prion-like mechanism based on the self-templating of its amyloid conformation. This prion-like aggregation could explain two key aspects of synucleinopathies: the invasion of different brain regions by the pathological inclusions during disease progression (Braak hypothesis) and the variety of clinical presentations associated with the aggregation of a single protein, a variety that could be linked to the polymorphism of the amyloid forms of alpha-synuclein.
Our team focuses on the structural, molecular, and cellular bases of the replicative and neuro-invasive processes of synucleinopathies, employing multiple approaches:
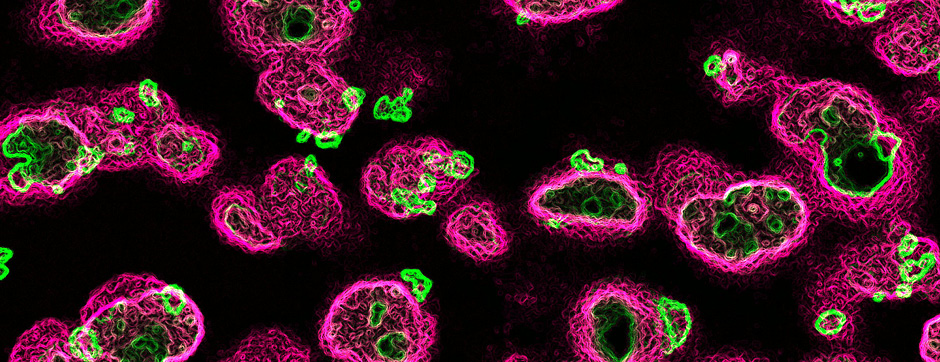
- Biochemistry: Production and analytical biochemistry are used to produce, isolate, and characterize amyloid complexes, providing structure-function information that can shed light on the molecular mechanisms promoting the progression of pathological amyloid aggregation and identify pharmacologically targetable sites in the assemblies.
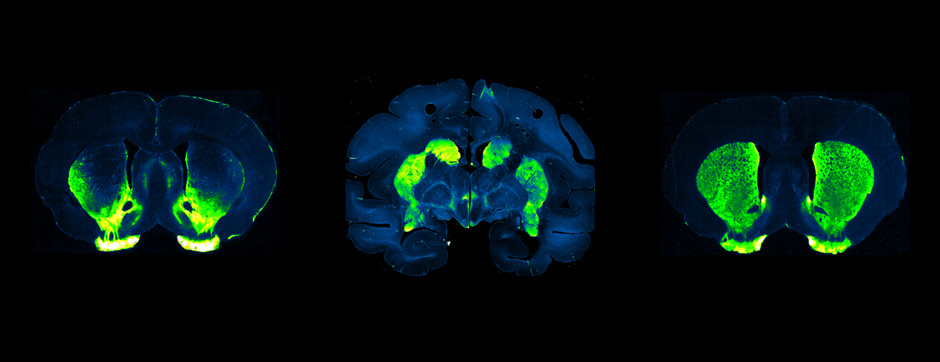
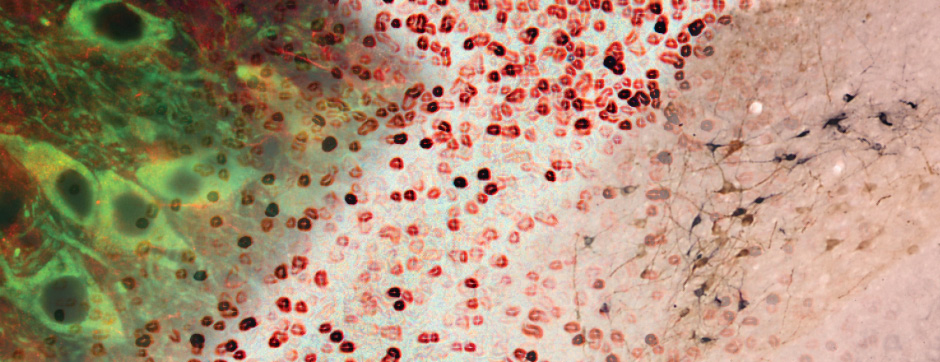
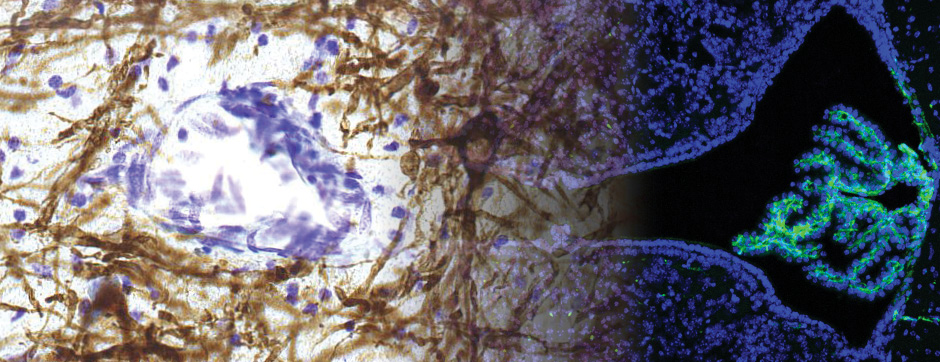
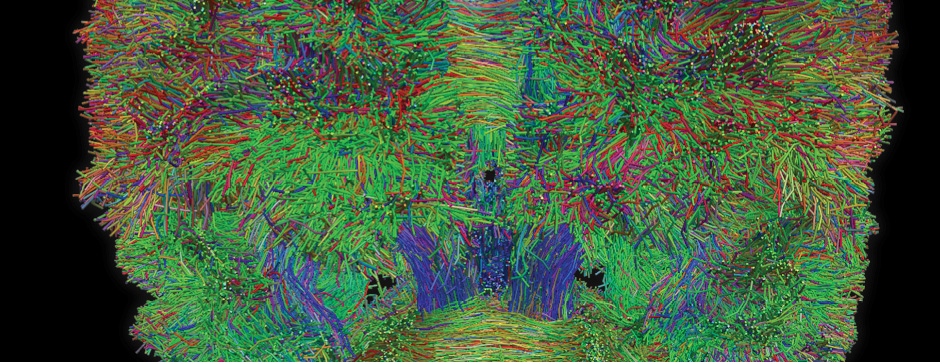
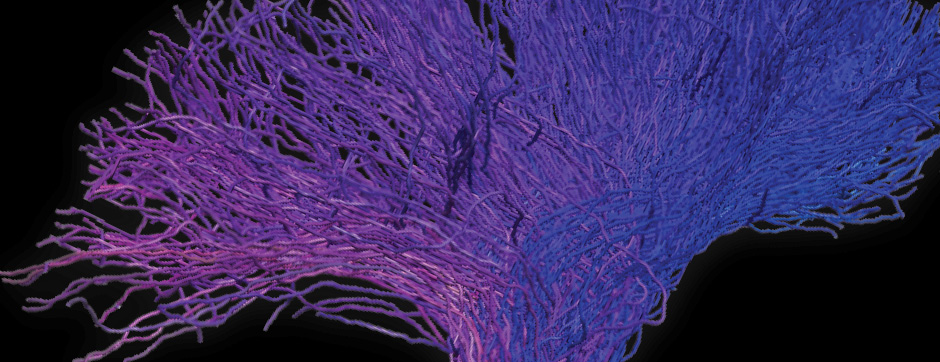
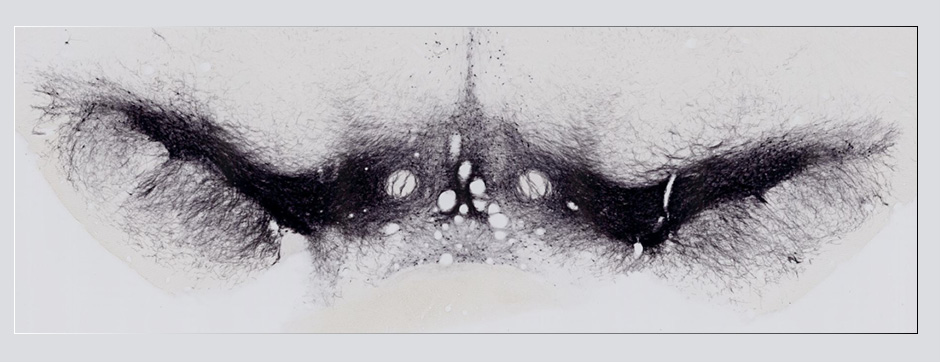
- Models based on primary cultures of mouse cortical neurons (high content analysis) and in vivo models (stereotaxic intracerebral injections) with exposure to alpha-synuclein preformed fibrils (PFFs) allow us to explore the propagation phenomenon and analyze the cellular processes involved in the fate of the neo-formed aggregates, the cell types involved, and the pathways activated by neurons or oligodendrocytes in response to the experimental induction of aggregation.
- Methodologies for quantifying histological markers using image analysis allow a quantitative study of pathology progression in vivo and the evaluation of the effects of pharmacological modulations.
With the use of these experimental and analytical methodologies, our team is particularly interested in studying the molecular aspects that differentiate PD and MSA and the downstream cellular mechanisms dealing with the aggregation process. These mechanisms could, on one hand, be involved in the origin of MSA (accumulation of synuclein in oligodendrocytes) and, on the other hand, determine neuronal dysfunction and loss.