





Simone BIDO
New insights into the circuitry underlying levodopa-induced dyskinesia in rodent models of Parkinson's disease
mars 2012 Directeur(s) de thèse : Michele MORARI Résumé de thèseAbnormal involuntary movements (AIMs) or dyskinesias are probably the most debilitating side-effect elicited by levodopa pharmacotherapy of Parkinson’s disease. Development of levodopa-induced dyskinesias (LID) reflects a processes of sensitization to levodopa taking place primarily in striatum, and leading to the abnormal response to dopaminomimetics. Despite the growing knowledge about the intracellular pathways involved in the development of LID, little is known about the impact of antidyskinetic treatments on the basal ganglia circuitry. In the present thesis, we used microdialysis to investigate the neurochemical and behavioural changes exerted by different antidyskinetic treatments or approaches in basal ganglia. We first found that levodopa evoked AIMs, and simultaneously elevated GABA levels in the substantia nigra reticulata but not globus pallidus of dyskinetic mice and rats, suggesting the involvement of the striato-nigral “direct” GABAergic pathway in both species (Bido et al., J Neurochem 118, 1043-1055, 2011). Amantadine (the only antidyskinetic drug marketed for treating LID) attenuated AIMs expression and prevented the nigral GABA rise (Bido et al., J Neurochem 118:1043-55, 2011), suggesting nigral GABA as a neurochemical correlate of LID. To further investigate which pathway is involved in LID and in the antidyskinetic effect of amantadine, we took advantage from recent studies showing the specificity of Ras-guanine nucleotide-releasing factor 1 and 2 to selectively couple NR2B and NR2A NMDA receptor subunits, respectively. We showed that blockade of striatal expression of Ras-GRF1 using a lentiviral vector carrying a short hairpin RNA (LV Ras-GRF1) caused an attenuation of LID development and expression, which was accompanied by the lack of the increase in nigral GABA. However, in LV Ras-GRF1 mice the antidyskinetic effect of amantadine and its neurochemical correlates were lost, suggesting LV Ras-GRF1 might interfere with the antidyskinetic effect of amantadine by acting on the same target (possibly the NR2B receptor). Conversely, injection of a viral construct expressing a small hairpin directed against RasGRF2 caused only a not significant reduction of LID, and did not prevent the increase of nigral GABA following L-DOPA. In these mice, also the antidyskinetic effect of amantadine remained unaltered (Bido et al., in preparation).
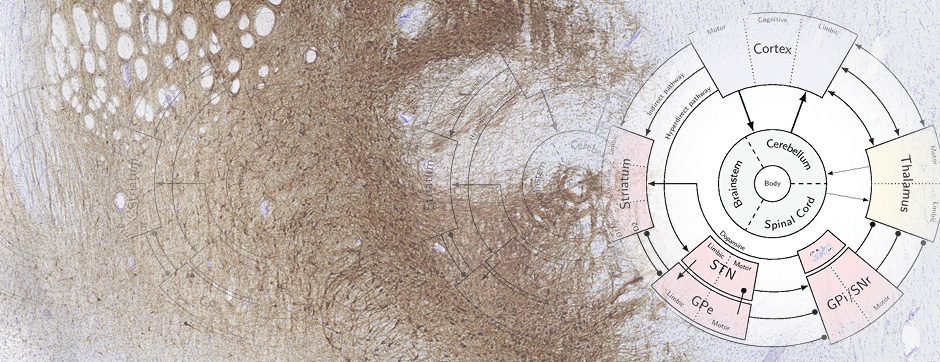
To confirm the involvement of the direct pathway in LID, and dissect out the role of striatal and nigral dopamine D1 and D2 receptors we performed regional perfusion (striatum and substantia nigra pars reticulata) of selective D1 and D2 antagonists simultaneously with systemic L-DOPA administration. Intrastriatal blockade of D1 receptor attenuated LID and prevented the accompanying rise of nigral GABA levels whereas blockade of D2 receptor was ineffective (Mela et al., Neurobiol Dis 45, 574-583, 2012). When perfused in the substantia nigra, both the D1 and D2 antagonists attenuated LID expression, although only the D1 antagonist prevented the GABA rise.
Overall, the data provide neurochemical evidence that LID is accompanied by activation of D1-receptor expressing striato-nigral GABAergic neurons, and that the antidyskinetic effect of amantadine partly relies on the modulation of this pathway, possibly through NR2B-subunit expressing NMDA receptors. Nonetheless, by using different antidyskinetic approaches we were able to cause only ~50% reduction of LID in face of a complete inhibition of the GABA rise in substantia nigra. This points to the existence of other important neurochemical modulators of LID, possibly also in brain structures outside the basal ganglia.

































































































